Research
Transcriptional regulation by the retinoblastoma family corepressors. Retinoblastoma proteins regulate cell cycle genes in most eukaryotes, and the retinoblastoma pathway is commonly disrupted in human cancer. In some metazoans, the genes for retinoblastoma proteins have duplicated; mammals have three paralogs, while Drosophila, unique among the insects, has two. Our studies of Drosophila Rbf1 (with the laboratory of R. William Henry, MSU) revealed an “instability element” in the C-terminus that plays a critical role in the activity and turnover of this protein. Interestingly, this element is conserved in human RB homologs p107 and p130, but it appears to have undergone evolutionary changes in RB itself, as well as Drosophila Rbf2.
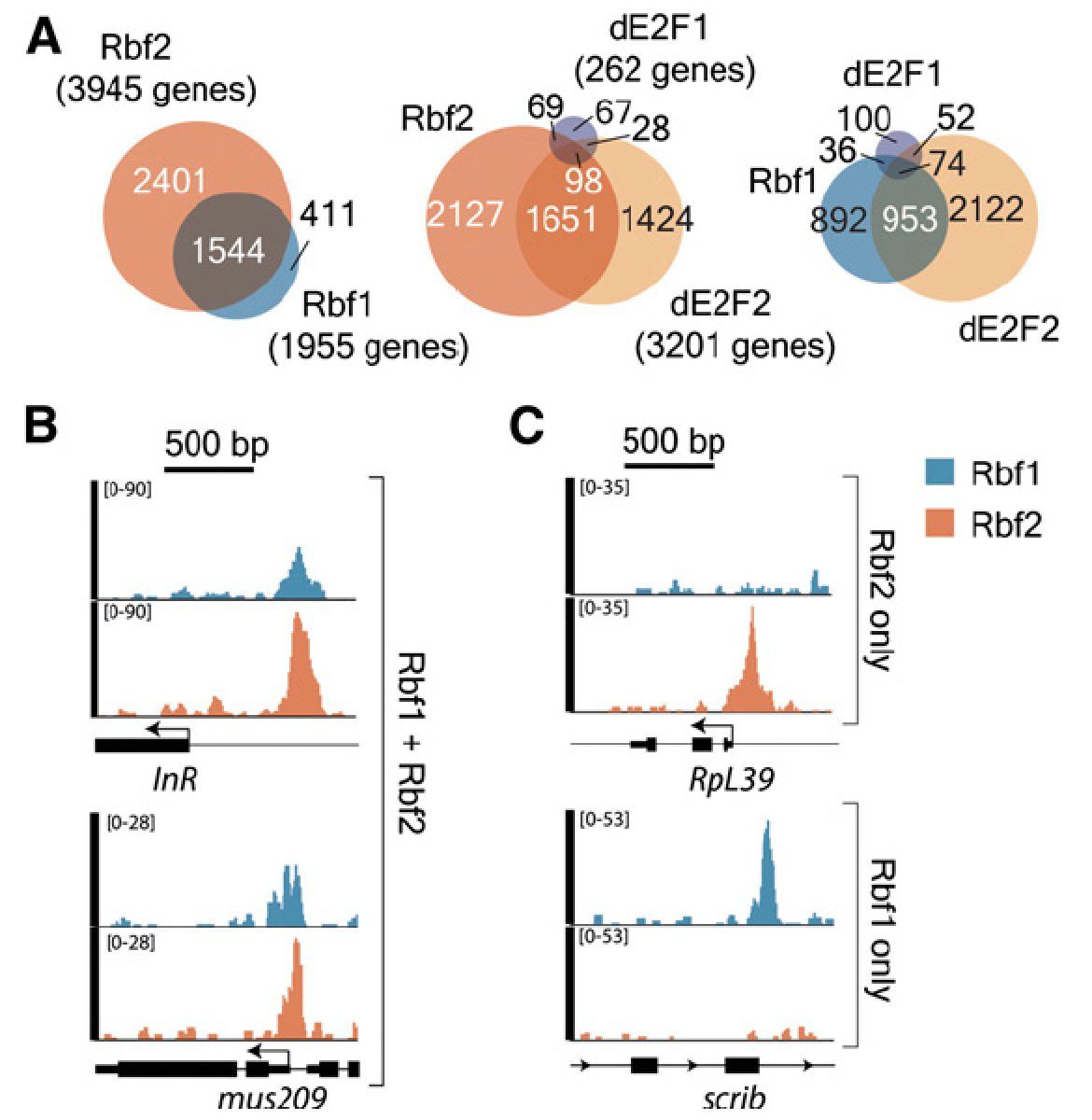 Our genome-wide analysis of Rbf1 and Rbf2 binding revealed that these proteins have
only partially overlapping occupancy, suggesting that they have different functional
roles. One likely role for Rbf2 in particular is the regulation of ribosomal protein
genes, generally regarded as “housekeeping” genes, but in fact tightly regulated in
response to developmental and nutritional cues. Disruption of RB pathways may thus
impact not only cell cycle, but also cell growth. ‘Omics analysis of Rbf1 and Rbf2
function (in collaboration with Philipp Kapranov, Huaqiao University) has revealed
multiple pathways in cellular signaling and growth control influenced by Rbf proteins.
Our genome-wide analysis of Rbf1 and Rbf2 binding revealed that these proteins have
only partially overlapping occupancy, suggesting that they have different functional
roles. One likely role for Rbf2 in particular is the regulation of ribosomal protein
genes, generally regarded as “housekeeping” genes, but in fact tightly regulated in
response to developmental and nutritional cues. Disruption of RB pathways may thus
impact not only cell cycle, but also cell growth. ‘Omics analysis of Rbf1 and Rbf2
function (in collaboration with Philipp Kapranov, Huaqiao University) has revealed
multiple pathways in cellular signaling and growth control influenced by Rbf proteins.
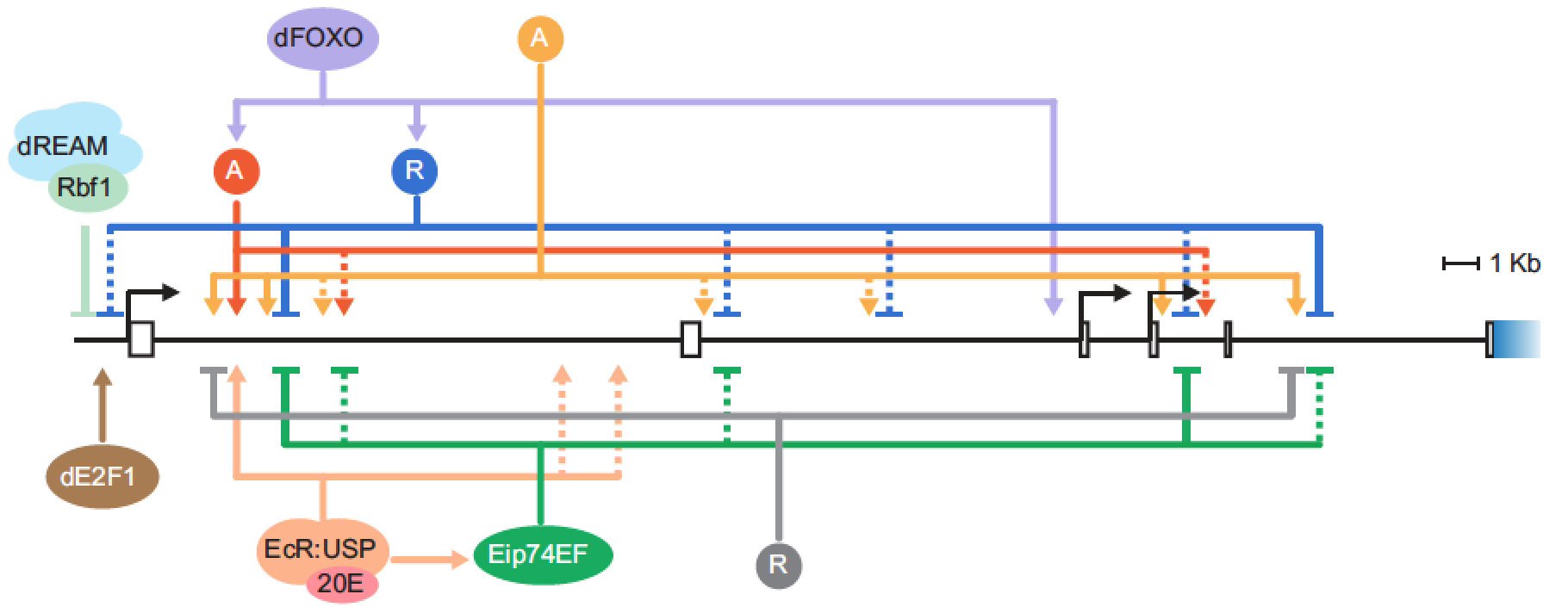
Transcriptional regulation of the insulin receptor gene. Our interest in retinoblastoma target genes led to consideration of genes involved in insulin signaling; a large fraction of the genes for components of this pathway are direct Rbf1 targets. Of particular interest is the insulin receptor gene, which is widely expressed and was originally described as a “housekeeping gene”. Our research reveals that this gene has sophisticated, complex regulatory inputs from multiple enhancers located throughout some 30 kbp of intronic sequence. Transcriptional regulation of this gene in humans is found to respond sensitively to developmental and environmental signals, but the physiological consequence of such variation is unknown. Our system will permit us to identify the importance of cis regulatory variation found at a population and species level, and ultimately determine the significance of SNPs in the human gene linked to disease. Experimental dissection of this gene’s enhancers is combined with bioinformatic analysis of the locus (in collaboration with the laboratory of Jianrong Wang, MSU).
Chromatin mechanisms of transcriptional control. Transcriptional enhancers are the key mediators of cellular signaling for control
of developmentally regulated genes. Genome-wide studies have associated particular
chromatin structures and histone protein modifications with active and inactive enhancers.
We identified the molecular mechanisms associated with two strikingly different classes
of transcriptional repressor, “short-range” repressors such as Knirps that locally
inhibit action of activators, and “long-range” repressors such as Hairy that can inhibit
activators bound to enhancers several kbp away. 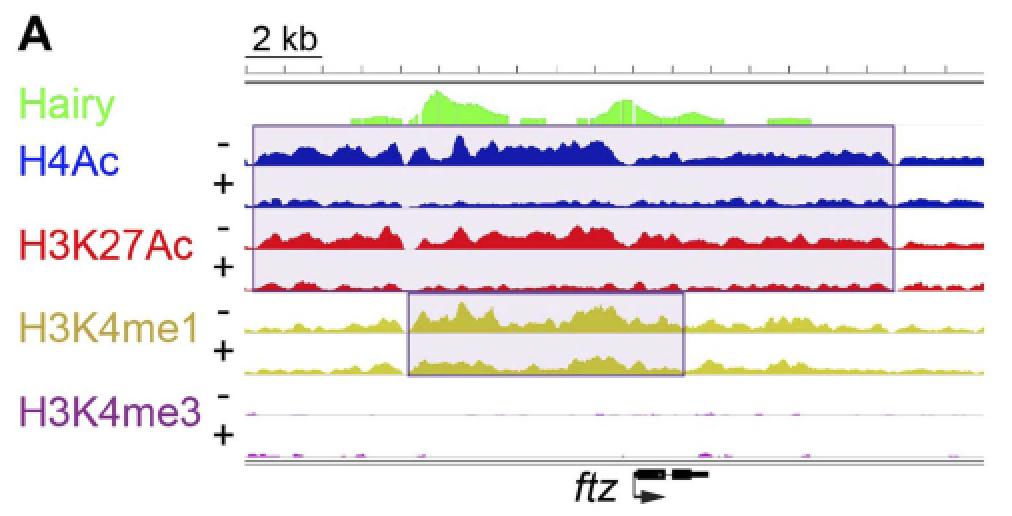 Knirps remodels chromatin structure very locally, likely putting it in an inaccessible
form for activators, while Hairy coordinates a suite of histone protein modifications
that involve loss of acetylation and methylation marks. Interestingly, while Hairy
impacts the chromatin states of large tracts of the genome, many of these impacts
are apparently “off target” – fortunately the transient modifications can be readily
reversed when Hairy goes away. We propose that these genome-wide off-target changes
are a fertile ground for evolution of novel regulatory sequences.
Knirps remodels chromatin structure very locally, likely putting it in an inaccessible
form for activators, while Hairy coordinates a suite of histone protein modifications
that involve loss of acetylation and methylation marks. Interestingly, while Hairy
impacts the chromatin states of large tracts of the genome, many of these impacts
are apparently “off target” – fortunately the transient modifications can be readily
reversed when Hairy goes away. We propose that these genome-wide off-target changes
are a fertile ground for evolution of novel regulatory sequences.
Cracking the code of cis-regulatory grammar. The transcriptional output of regulatory regions is dictated by the composition
of transcription factor binding sites, as well as chromatin occupancy and cellular
signaling. Heritable human diseases and conditions are caused by loss or gain of a
single binding site, yet predicting these effects remains very difficult, due to the
flexible nature of cis-regulatory information (many different sequences can encode
the same output, and similar sequences can have very different outputs). 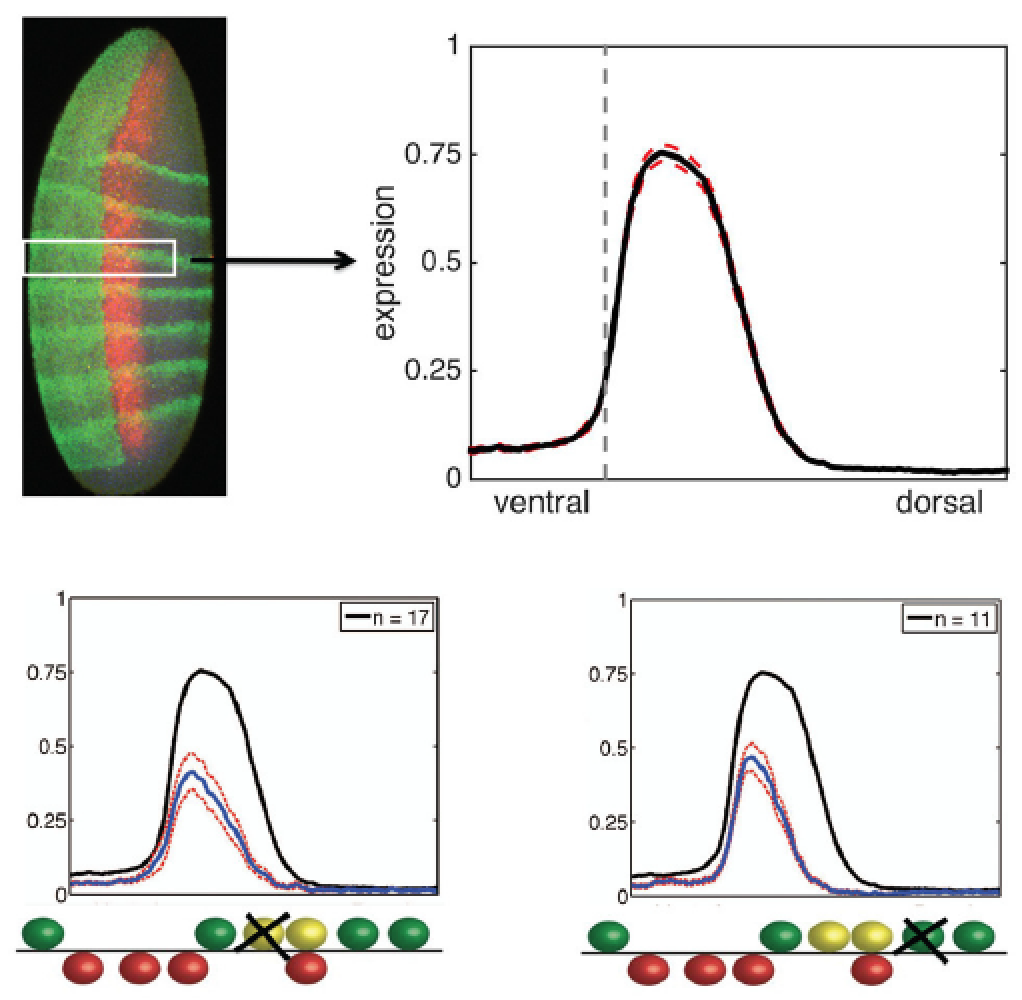 We showed that subtle changes in sequence can produce dramatic effects in the output
of an enhancer; for instance, small changes in the spacing of a short-range repressor
to neighboring factors can “tune” an enhancer to respond strongly or weakly to the
repressor. Quantitative, systematic analysis of the rhomboid enhancer input/output function allows us to fit parameters to models that describe
how transcription factors binding to this enhancer can cooperate, or antagonize one
another in complex fashions. The predictive power of such models remains to be fully
tested; ultimately we hope to be smarter than a single cell (!), which can effortlessly
interpret the cis regulatory code of diverse enhancers introduced into the nucleus.
We showed that subtle changes in sequence can produce dramatic effects in the output
of an enhancer; for instance, small changes in the spacing of a short-range repressor
to neighboring factors can “tune” an enhancer to respond strongly or weakly to the
repressor. Quantitative, systematic analysis of the rhomboid enhancer input/output function allows us to fit parameters to models that describe
how transcription factors binding to this enhancer can cooperate, or antagonize one
another in complex fashions. The predictive power of such models remains to be fully
tested; ultimately we hope to be smarter than a single cell (!), which can effortlessly
interpret the cis regulatory code of diverse enhancers introduced into the nucleus.